

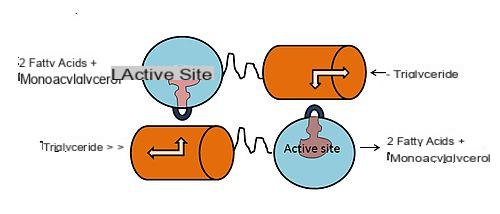
A lipoproteína lipase (LPL) é uma enzima encontrada nas células endoteliais que revestem a superfície interna dos capilares sanguíneos. Está particularmente concentrado ao nível do endotélio capilar do tecido muscular esquelético, tecido cardíaco e tecido adiposo. Não surpreendentemente, a função da lipase de lipoproteína é hidrolisar os triglicerídeos contidos nas lipoproteínas (quilomícrons e VLDL), liberando dois ácidos graxos livres e um monoacilglicerol. Os produtos derivados desta hidrólise de triglicéridos difundem-se nas células, onde podem essencialmente cumprir dois destinos: o primeiro deve ser metabolizado no músculo esquelético e no coração, o segundo, típico de períodos de alimentação excessiva (excesso de energia), é para serem usados como substratos para a ressíntese de triglicerídeos, então acumulados como reserva de energia.
A insulina aumenta a expressão da lipase lipoproteica no tecido adiposo branco, favorecendo a hidrólise dos triglicerídeos sanguíneos em glicerol e ácidos graxos; assim, este último pode entrar nos adipócitos e então ser reesterificado com glicerol, formando triglicerídeos de reserva.
Deficiência familiar de lipoproteína lipase (doença de Burger-Grutz ou hiperlipoproteinemia familiar tipo I)
Doença autossômica recessiva, com incidência igual a um caso em 100.000 pessoas. Ela ocorre em indivíduos homozigotos para uma mutação no gene que codifica a lipase da lipoproteína. A conseqüente deficiência desta enzima faz com que os indivíduos afetados por esta doença apresentem níveis particularmente elevados de triglicerídeos (geralmente excedendo 800-1000 mg / dL), devido ao bloqueio do metabolismo dos quilomícrons. A hipertrigliceridemia grave é acompanhada, desde a infância, por maior incidência de pancreatite, dor abdominal, xantomas eruptivos (pápulas amareladas com contornos avermelhados distribuídos nas regiões do corpo sob pressão) e hepatoesplenomegalia (aumento anormal do fígado e baço). O aumento do risco cardiovascular também é sensível, embora a retinopatia às vezes esteja presente.
Deficiência familiar de APO-C2
Uma proteína muito importante, por ser capaz de ativar a lipoproteína lipase, é a Apo-lipo-proteína-C2 ou APO-C2. A deficiência dessa proteína, expressa na superfície de VLDL e quilomícrons, pode causar hiperlipoproteinemia caracterizada por hipertrigliceridemia (triglicerídeos elevados no sangue). Consequentemente, a deficiência de APO-C2 está associada a um maior risco de aterosclerose precoce e pancreatite, mais comum na velhice. Também neste caso a doença está ligada a uma mutação autossômica recessiva, especificamente no gene que codifica para APO-C2.
Lipoproteinlipase, dieta, medicamentos e suplementos
A lipoproteína lipase ou deficiência de APO-C2 pode ser tratada com uma dieta pobre em gorduras, consumida em quantidades não superiores a 10-20 gramas por dia. As gorduras de cadeia intermediária, que se ligam diretamente à albumina e não exploram os quilomícrons para serem transportadas na corrente sanguínea, serão claramente preferidas. Ao mesmo tempo, é necessário abolir o álcool e garantir um suprimento adequado de vitaminas lipossolúveis e ácidos graxos essenciais. Os ômega-3 de origem em peixes (EPA e DHA), em particular, mostraram propriedades notáveis na redução de hipotiglicerídeos e, como tal, são usados em altas doses para reduzir os níveis de triglicerídeos. Outros fármacos com atividade semelhante são os fibratos e o ácido nicotínico, que exercem sua ação redutora de hipotiglicerídeos também por aumentar a expressão da enzima lipoproteína lipase.