


Active principles: Strontium (strontium ranelate)
PROTELOS 2 g granules for oral suspension
Why is Protelos used? What is it for?
PROTELOS is a medicine used to treat severe osteoporosis:
- in postmenopausal women
- in adult men
at high risk of fractures, for which it is not possible to resort to alternative treatments. In postmenopausal women, strontium ranelate reduces the risk of spine and hip fractures.
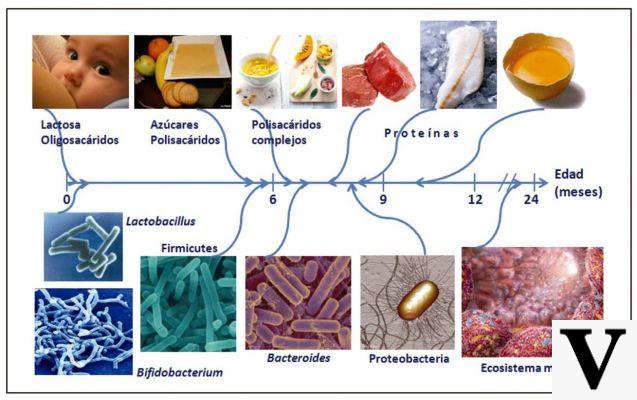
Osteoporosis
The body continually destroys old bone and forms new bone. In the case of osteoporosis, the body destroys more bone than is formed, so that gradually bone loss occurs and the bones become thinner and more fragile. This is especially the case in postmenopausal women.
Many people with osteoporosis have no symptoms, and it is possible not to even know that you have osteoporosis. However, osteoporosis predisposes to fractures (broken bones) especially in the spine, hips and wrists.
How PROTELOS works
PROTELOS, which contains the active substance strontium ranelate, belongs to a group of medicines used to treat bone diseases. PROTELOS reduces bone destruction and stimulates bone reconstruction, thereby reducing the risk of fractures. The new bone formed is of normal quality.
Contraindications When Protelos should not be used
Do not fasten PROTECT THEM
- if you are allergic to strontium ranelate or any of the other ingredients of PROTELOS (listed in section 6).
- if you have or have ever had a thrombosis (for example, affecting the blood vessels in the leg or lungs).
- if you are immobilized permanently or for a certain period, such as if you are in a wheelchair, or if you are bedridden or if you need to have surgery or if you are in post-operative recovery. The risk of venous thrombosis (thrombosis in the leg or lung) may be higher in the case of prolonged immobilization.
- if you have known ischemic heart disease, or cerebrovascular disease, eg. if you have been diagnosed with a heart attack, stroke, or transient ischemic attack (temporary reduction in blood flow to the brain; also known as a "mini-stroke"), angina, or a blockage of blood vessels in the heart or brain.
- if you have or have had problems with your blood circulation (peripheral arterial disease) or if you have had surgery on the arteries in your legs.
- if you have high blood pressure that is not controlled by treatment.
Precautions for use What you need to know before taking Protelos
Talk to your doctor or pharmacist before taking PROTELOS:
- if you are at risk of heart disease; this includes high blood pressure, high cholesterol, diabetes, smoking
- if you are at risk for thrombosis
- if you have severe kidney disease.
Your doctor will evaluate the condition of your heart and blood vessels at regular intervals, usually every 6-12 months, throughout the period of treatment with PROTELOS.
During treatment, if you experience an allergic reaction (such as swelling of the face, tongue or throat, difficulty breathing or swallowing, skin rash), you should immediately stop taking PROTELOS and contact your doctor (see section 4). Potentially life-threatening skin rashes (Stevens-Johnson Syndrome (SJS), toxic epidermal necrolysis and severe hypersensitivity reactions (DRESS)) have been reported while using PROTELOS. The greatest risk of incidence of severe skin reactions is within the first few weeks of treatment for Stevens-Johnson syndrome and toxic epidermal necrolysis and generally around 3-6 weeks for DRESS. If you develop a rash or serious skin symptoms (see section 4), stop taking PROTELOS, contact your doctor immediately and report that you are taking this medicine. If you have experienced Stevens-Johnson syndrome, toxic epidermal necrolysis or DRESS while using PROTELOS, you should never restart PROTELOS treatment. If you are of Asian descent, talk to your doctor before taking PROTELOS as you may have a higher risk of skin reactions.
Children and adolescents
PROTELOS is not indicated for use in children and adolescents (under 18 years of age).
Interactions Which drugs or foods can change the effect of Protelos
Other medicines and PROTELOS
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines.
Stop taking PROTELOS if you need to take oral tetracyclines such as doxycycline or quinolones such as ciprofloxacin (two types of antibiotics). You can restart PROTELOS when you have finished taking these antibiotics. If you are not sure, ask your doctor or pharmacist. If you are taking medicines containing calcium, allow at least 2 hours to elapse before taking PROTELOS.
If you take antacids (medicines to relieve heartburn), take them at least 2 hours after taking PROTELOS. If this is not possible, taking the two medicines at the same time is acceptable.
If it is necessary to test the level of calcium in the blood or urine, you must inform the laboratory that you are taking PROTELOS, as it may interfere with some test methods.
PROTELOS with food and drink
Food, milk and its derivatives reduce the absorption of strontium ranelate. It is recommended to take PROTELOS between meals, preferably at bedtime, at least two hours after taking food, milk and dairy products or calcium supplements.
Warnings It is important to know that:
Pregnancy and breastfeeding
Do not take PROTELOS during pregnancy or when you are breast-feeding. In case of accidental use during pregnancy or breastfeeding, stop taking the medicine immediately and inform your doctor.
Driving and using machines
PROTELOS is unlikely to affect the ability to drive or use machines.
PROTELOS contiene aspartame(E951)
If you have phenylketonuria (a rare inherited disorder of metabolism), consult your doctor before starting to take this medicine.
Dose, Method and Time of Administration How to use Protelos: Posology
Treatment should only be initiated by a physician experienced in the treatment of osteoporosis.
Always take this medicine exactly as your doctor or pharmacist has told you. If in doubt, consult your doctor or pharmacist.
PROTELOS is for oral use. The recommended dose is one 2g sachet per day.
It is recommended to take PROTELOS at bedtime, preferably at least 2 hours after dinner. You can also go to bed immediately after taking PROTELOS if you wish.
Take the granules contained in the sachets after having suspended it in a glass containing at least 30 ml of water (approximately one third of a standard glass) (see instructions below). PROTELOS can interact with milk and its derivatives; therefore it is important that PROTELOS is only mixed with water in order to be sure that the medicine works correctly.
- Pour the granules from the sachet into a glass;
- Add water;
- Stir until the granulate is completely dispersed in the water.
Drink immediately. Do not allow more than 24 hours to pass before drinking the suspension. If for some reason you cannot take the medicine right away, remember to mix it again before drinking.
Your doctor may advise you to take calcium and vitamin D supplements in addition to PROTELOS. Do not take calcium supplements at bedtime, at the same time as PROTELOS.
Your doctor will tell you how long to continue taking PROTELOS. Osteoporosis therapy usually takes a long time. It is important to continue taking PROTELOS as long as your doctor prescribes it.
Overdose What to do if you have taken too much Protelos
If you take more PROTELOS than you should
If you take more PROTELOS sachets than your doctor has prescribed, please tell your doctor or pharmacist. They may advise you to drink milk or take antacids to reduce the absorption of the active substance.
If you forget to take PROTELOS
Do not take a double dose to make up for a forgotten dose. Simply take the next dose at the appointed time.
If you stop taking PROTELOS
It is important to continue taking PROTELOS for as long as your doctor has prescribed. PROTELOS can treat severe osteoporosis only if it is taken continuously. If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
Side Effects What are the side effects of Protelos
Like all medicines, this medicine can cause side effects, although not everybody gets them.
Stop taking PROTELOS and tell your doctor right away if you experience any of the following side effects:
Common (may affect up to 1 in 10 patients):
- Heart attack: Sudden squeezing pains in the chest that can extend to the left arm, jaw, stomach, back and / or shoulders. Other symptoms may be: nausea / vomiting, sweating, shortness of breath, palpitations, (extreme) fatigue and / or dizziness. In patients at high risk for heart disease, a heart attack can occur with a common frequency. If you are a high-risk patient, your doctor will not prescribe PROTELOS for you.
- Blood clots in the veins (thrombosis): pain, redness, swelling of the legs, sudden chest pain or difficulty in breathing.
Rare (may affect up to 1 in 1.000 patients):
- Signs of severe hypersensitivity reactions (DRESS): initially as flu-like symptoms and rash on the face, then extended rash with a high temperature (uncommon), increase in liver enzyme levels found in blood tests (uncommon), increase in a certain type of white blood cells (eosinophilia) (rare) and enlarged lymph nodes (uncommon).
Very rare (may affect up to 1 in 10.000 patients):
- Signs of potentially life-threatening skin rash (Stevens-Johnson syndrome, toxic epidermal necrolysis): Initially as reddish target-like patches or circular patches often with central blisters on the trunk. Additional signs may include ulceration of the mouth, throat, nose, genitals and conjunctivitis (red and swollen eyes). These potentially life-threatening skin rashes are often accompanied by flu-like symptoms. The rash may progress to blistering all over the body or peeling of the skin.
Other possible side effects
Very common (may affect more than 1 in 10 patients):
Itching, hives, skin rash, angioedema (such as swelling of the face, tongue or throat, difficulty breathing or swallowing), pain in bones, limbs, muscles and / or joints, muscle cramps.
Common
Vomiting, abdominal pain, reflux, difficulty digesting, constipation, flatulence, difficulty sleeping, inflammation of the liver (hepatitis), swelling of the limbs, bronchial hyperreactivity (symptoms include wheezing, shortness of breath and cough), increased level of an enzyme muscle (creatine phosphokinase). Nausea, diarrhea, headache, eczema, memory impairment, fainting, tingling, dizziness, vertigo. However, these effects are mild and transient and usually do not require discontinuation of treatment. Tell your doctor if any of these side effects become troublesome or persistent.
Uncommon (may affect up to 1 in 100 patients):
(Convulsions, irritation of the oral mucosa (such as mouth ulcerations and inflammation of the gums), hair loss, confusion, feeling sick, dry mouth, skin irritation.
Rare:
Reduced production of blood cells in the bone marrow. If you have stopped therapy due to hypersensitivity reactions, you should not restart PROTELOS.
Reporting of side effects
If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. You can also report side effects directly via the national reporting system listed in Appendix V. By reporting side effects you can help provide more information on the safety of this medicine.
Expiry and Retention
Keep this medicine out of the sight and reach of children.
Do not use this medicine after the expiry date which is stated on the carton and sachet after EXP. The expiry date refers to the last day of that month.
This medicine does not require any special storage conditions.
Once reconstituted in water, the suspension is stable for 24 hours. However, it is recommended to drink the suspension immediately after preparation (see section 3).
Do not throw any medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. This will help protect the environment.
Composition and pharmaceutical form
Thing contains PROTELOS
- The active ingredient is strontium ranelate. Each sachet contains 2 g of strontium ranelate.
- The other ingredients are aspartame (E 951), maltodextrin, mannitol (E 421).
What PROTELOS looks like and contents of the pack
PROTELOS is available in sachets containing a yellow granules for oral suspension. PROTELOS is supplied in packs of 7, 14, 28, 56, 84 or 100 sachets. Not all pack sizes may be marketed.
Learn more about Protelos are available in the "Summary of Characteristics" tab.01.0 NAME OF THE MEDICINAL PRODUCT
PROTELOS 2 G
▼ Medicinal product subject to additional monitoring. This will allow the rapid identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions See section 4.8 for information on how to report adverse reactions.
02.0 QUALITATIVE AND QUANTITATIVE COMPOSITION
Each sachet contains 2 g of strontium ranelate.
Excipient with known effect: each sachet also contains 20 mg of aspartame (E 951).
For the full list of excipients, see section 6.1.
03.0 PHARMACEUTICAL FORM
Granules for oral suspension.
Yellow granulate.
04.0 CLINICAL INFORMATION
04.1 Therapeutic indications
Treatment of severe osteoporosis:
- in postmenopausal women
- in adult men
at high risk of fractures, for which treatment with other medicines approved for the treatment of osteoporosis is not possible due, for example, to contraindications or intolerance.
Strontium ranelate reduces the risk of vertebral and hip fractures in postmenopausal women (see section 5.1).
The decision to prescribe strontium ranelate should be based on an assessment of the individual patient's overall risks (see sections 4.3 and 4.4).
04.2 Posology and method of administration
Treatment should only be initiated by a physician experienced in the treatment of osteoporosis.
dosage
The recommended dose is one 2 g sachet once daily for oral administration.
Due to the nature of the condition being treated, strontium ranelate is intended for long-term use.
Absorption of strontium ranelate is reduced by food, milk and its derivatives and therefore PROTELOS should be administered between meals. Due to its slow absorption, PROTELOS should be taken at bedtime, preferably at least two hours after a meal (see sections 4.5 and 5.2).
Patients being treated with strontium ranelate should take vitamin D and calcium supplements if their dietary intake is insufficient.
Elderly patients
The efficacy and safety of strontium ranelate have been demonstrated in a large sample of adult men and postmenopausal women of all ages (up to 100 years at inclusion) with osteoporosis. No dosage adjustment is required in relation to age.
Patients with renal insufficiency
Strontium ranelate is not recommended in patients with severe renal impairment (creatinine clearance less than 30 ml / min) (see sections 4.4 and 5.2). No dosage adjustment is required in patients with mild to moderate renal impairment (creatinine clearance 30 - 70 ml / min) (see sections 4.4 and 5.2).
Patients with hepatic insufficiency
No dosage adjustment is required in patients with hepatic insufficiency (see section 5.2).
Pediatric population
The safety and efficacy of PROTELOS in children aged below 18 years have not been established.
No data are available.
Method of administration
For oral use.
The granules of the sachets should be taken after suspension in a glass containing a minimum of 30 ml of water (approximately one third of a normal glass).
Although studies related to use have shown that strontium ranelate remains stable in suspension for 24 hours after preparation, the suspension should be drunk immediately after its preparation.
04.3 Contraindications
- Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
- Current or previous venous thromboembolism (VTE), including deep vein thrombosis and pulmonary embolism.
- Temporary or permanent immobilization due for example to surgery or a prolonged stay in bed.
- Known, current or previous ischemic heart disease, peripheral arterial disease and / or cerebrovascular disease.
- Uncontrolled hypertension.
04.4 Special warnings and appropriate precautions for use
Ischemic cardiac events
In a pooled analysis of placebo-controlled randomized clinical trials in postmenopausal osteoporotic patients, a significant increase in myocardial infarction was observed in patients treated with PROTELOS compared to those treated with placebo (see section 4.8).
Patients should be evaluated for cardiovascular risk before starting treatment.
Patients with significant risk factors for cardiovascular events (e.g. hypertension, hyperlipidaemia, diabetes mellitus, smoking) should only be treated with strontium ranelate after careful consideration (see sections 4.3 and 4.8).
During treatment with PROTELOS, these cardiovascular risks should be monitored at regular intervals, usually every 6-12 months.
Treatment should be discontinued if the patient develops ischemic heart disease, peripheral arterial disease, cerebrovascular disease or if hypertension is not controlled (see section 4.3).
venous thromboembolism
In phase III placebo-controlled studies, treatment with strontium ranelate was associated with an increased annual incidence of venous thromboembolism (VTE), including pulmonary embolism (see section 4.8). The cause of this increase is unknown. PROTELOS is contraindicated in patients with previous venous thromboembolism (see section 4.3) and should be used with caution in patients at risk of VTE.
During the treatment of patients over 80 years of age at risk of VTE, the need for continued treatment with PROTELOS should be reassessed. Treatment with PROTELOS should be stopped as soon as possible in the event of an illness or condition leading to immobilization (see section 4.3) and adequate preventive measures should be taken. Therapy should not be resumed until the condition that led to the immobilization has resolved and the patient is completely mobile. When a VTE occurs, PROTELOS should be discontinued.
Use in patients with renal insufficiency
In the absence of bone safety data in patients with severe renal insufficiency treated with strontium ranelate, PROTELOS is not recommended in patients with a creatinine clearance below 30 ml / min. (see section 5.2). In accordance with good clinical practice, periodic monitoring of renal function is recommended in patients with chronic renal failure. Continuation of PROTELOS therapy in patients who develop severe renal insufficiency should be evaluated on an individual basis.
Skin reactions
Life-threatening skin reactions (Stevens-Johnson Syndrome (SJS), toxic epidermal necrolysis (NET) and drug rash with eosinophilia and systemic symptoms (DRESS)) have been reported while using PROTELOS.
Patients should be informed of the signs and symptoms and carefully monitored for skin reactions. The greatest risk of incidence for SJS or NET is within the first few weeks of treatment and within 3-6 weeks for DRESS.
If signs and symptoms of SJS or NET (e.g., progressive skin rash often with blistering and mucosal lesions) or DRESS (e.g. rash, fever, eosinophilia and systemic involvement (e.g. adenopathy, hepatitis, nephropathy and lung disease) occur interstitial), PROTELOS treatment should be stopped immediately.
The best results in the management of SJS, NET or DRESS are obtained following an early diagnosis and immediate discontinuation of any suspect drug. Early discontinuation of treatment is associated with a better prognosis. The clinical picture of DRESS resolved in most cases with the discontinuation of PROTELOS treatment and with the initiation of corticosteroid therapy when necessary. Recovery may be slow and relapses of the syndrome have been reported in some cases after discontinuation of corticosteroid therapy.
In patients who have developed SJS, NET or DRESS with the use of PROTELOS, PROTELOS therapy should no longer be restarted.
A higher, although still rare, incidence of hypersensitivity reactions including skin rash, SJS or NET has been reported in patients of Asian descent.
Interactions with laboratory tests
Strontium interferes with colorimetric methods for determining blood and urinary concentrations of calcium. Therefore, in clinical practice, inductively coupled plasma atomic emission spectrometry or atomic absorption spectrometry methods should be used to ensure accurate assessment of blood and urinary calcium concentrations.
Excipient
PROTELOS contains aspartame, a source of phenylalanine, which can be dangerous for patients with phenylketonuria.
04.5 Interactions with other medicinal products and other forms of interaction
Food, milk and its derivatives, and medicinal products containing calcium can reduce the bioavailability of strontium ranelate by approximately 60 - 70%. Therefore, the administration of PROTELOS and these products should be separated by at least two hours (see sections 4.2 and 5.2).
Since divalent cations can form a poorly absorbable complex with oral tetracyclines (e.g. doxycycline) and quinolone antibiotics (e.g. ciprofloxacin) at the gastrointestinal level, co-administration of strontium ranelate with these medicinal products is not recommended. As a precautionary measure, PROTELOS should be discontinued during treatment with oral tetracyclines or quinolone antibiotics.
An in vivo clinical study of drug interactions showed that the intake of aluminum and magnesium hydroxides, within the two hours prior to or at the same time as strontium ranelate, caused a slight decrease in the absorption of strontium ranelate (20-25% decrease). AUC), while absorption remained practically unaltered when the antacid was administered two hours after strontium ranelate. It is therefore preferable to take antacids at least two hours after taking PROTELOS. However, as it is recommended to take PROTELOS at bedtime when this dosing schedule is not applicable, concomitant intake remains acceptable.
No interaction was observed with oral vitamin D supplementation.
In clinical trials, no clinical interaction, nor a significant increase in blood strontium levels, has been demonstrated with medicinal products that, in current practice, are commonly prescribed concomitantly with PROTELOS, including: non-steroidal anti-inflammatory drugs ( including acetylsalicylic acid), anilides (such as paracetamol), H2 blockers and proton pump inhibitors, diuretics, digoxin and cardiac glycosides, organic nitrates and other vasodilators for heart disease, calcium-antagonists, beta-blockers, ACEs-inhibitors, angiotensin II antagonists, selective beta-2-adrenergic receptor agonists, oral anticoagulants, platelet aggregation inhibitors, statins, fibrates and benzodiazepine derivatives.
04.6 Pregnancy and breastfeeding
Pregnancy
Data from the use of strontium ranelate in pregnant women are not available. Animal studies have shown, at high doses, reversible bone effects in the offspring of rats and rabbits treated during pregnancy (see section 5.3). If PROTELOS is taken inadvertently during pregnancy, the treatment should be stopped.
Feeding time
Physico-chemical data suggest the excretion of strontium ranelate in breast milk. PROTELOS should not be used during breastfeeding.
Fertility
No effects on male and female fertility were observed in animal studies.
04.7 Effects on ability to drive and use machines
Strontium ranelate has no or negligible influence on the ability to drive or use machines.
04.8 Undesirable effects
Summary of the safety profile
PROTELOS has been studied in clinical trials involving approximately 8.000 people. Long-term safety was evaluated in phase III studies in postmenopausal women with osteoporosis treated for up to 60 months with strontium ranelate 2 g / day (n = 3.352) or placebo (n = 3.317). The mean age at the time of inclusion was 75 years and 23% of enrolled patients were between 80 and 100 years of age.
In a pooled analysis of randomized placebo-controlled trials in postmenopausal osteoporotic patients, the most common adverse reactions were nausea and diarrhea, generally reported at initiation of treatment, with no appreciable difference between groups in later stages. Discontinuation of therapy is mainly due to nausea. There was no difference in the nature of the adverse reactions between the treatment groups, regardless of whether the patients' age was less than or greater than 80 years at the time of inclusion.
Table of adverse reactions
The following adverse reactions have been reported during clinical trials and / or during post-marketing use of strontium ranelate. Adverse reactions are listed below, using the following convention: very common (≥1 / 10); common (≥1 / 100 to
| System and organ classification | Frequency | Adverse reaction |
| Disorders of the blood and lymphatic system | Uncommon | Lymphadenopathy (in association with skin hypersensitivity reactions) |
| Raro | Bone marrow insufficiency # | |
| Eosinophilia (in association with skin hypersensitivity reactions) | ||
| Metabolism and nutrition disorders | Common | Hypercholesterolemia |
| Psychiatric disorders | Common | Insomnia |
| Uncommon | Confusional state | |
| Nervous system disorders | Common | Headache |
| Disturbances of consciousness | ||
| Loss of memory | ||
| Dizziness | ||
| Paraesthesia | ||
| Uncommon | Seizures | |
| Ear and labyrinth disorders | Common | Dizziness |
| Cardiac pathologies | Common | Myocardial infarction |
| Vascular pathologies | Common | Venous thromboembolism (VTE) |
| Respiratory, thoracic and mediastinal disorders | Common | Bronchial hyperreactivity |
| Gastrointestinal disorders | Common | Nausea |
| Diarrhea and loose stools | ||
| Threw up | ||
| Abdominal pain | ||
| Gastrointestinal pain | ||
| Gastroesophageal reflux | ||
| Dyspepsia | ||
| Constipation | ||
| Flatulence | ||
| Uncommon | Oral mucosal irritation (stomatitis and / or mouth ulcerations) | |
| Dry mouth | ||
| Hepatobiliary disorders | Common | Hepatitis |
| Uncommon | Increase in serum transaminases (in association with skin hypersensitivity reactions) | |
| Skin and subcutaneous tissue disorders | Very common | Hypersensitivity skin reactions (rash, pruritus, urticaria, angioedema) § |
| Common | Eczema | |
| Uncommon | Dermatitis | |
| Alopecia | ||
| Raro | Adverse reactions with eosinophilia and systemic symptoms (DRESS) (see section 4.4) # | |
| Very rare | Serious skin adverse reactions (SCARs): Stevens-Johnson syndrome and toxic epidermal necrolysis * (see section 4.4) # | |
| Musculoskeletal and connective tissue disorders | Very common | Musculoskeletal pain (muscle spasm, myalgia, bone pain, arthralgia and pain in extremity) § |
| General disorders and administration site conditions | Common | Peripheral edema |
| Uncommon | Pyrexia (in association with skin hypersensitivity reactions) | |
| Malaise | ||
| Diagnostic tests | Common | Blood creatine phosphokinase (CPK) increased a |
§ The frequency in clinical trials was similar in the drug group and the placebo group.
* Reported as rare in Asian countries.
# For adverse reactions not observed in clinical trials, the upper limit of the 95% confidence interval is not greater than 3 / X with X representing the total sample size from all clinical trials and relevant studies.
a Musculoskeletal fraction> 3 times the upper limit of the normal range. In most cases, these values normalized spontaneously without any change in therapy.
Description of selected adverse reactions
venous thromboembolism
In phase III studies, the annual incidence of venous thromboembolism (VTE) events observed over 5 years was approximately 0,7% with a relative risk of 1,4 (95% CI = [1,0; 2,0, 4.4]) in patients treated with strontium ranelate versus placebo (see section XNUMX).
Myocardial infarction
In a pooled analysis of randomized placebo-controlled clinical trials in postmenopausal osteoporotic patients, a significant increase in myocardial infarction was observed in patients treated with strontium ranelate compared to patients given placebo (1,7% compared with at 1,1%), with a relative risk of 1,6 (95% CI = [1,07; 2,38]).
Reporting of suspected adverse reactions
04.9 Overdose
Symptoms
In a clinical study evaluating repeated administration of 4 g strontium ranelate daily for more than 25 days in healthy postmenopausal women, good tolerability was found. Single administration of doses up to 11 g to healthy young male volunteers did not cause any particular symptoms.
Management
No clinically relevant effects have been observed from the observation of overdose episodes during clinical trials (up to 4 g / day for a maximum time period of 147 days).
The administration of milk or antacids may be useful in reducing the absorption of the active ingredient. In the event of a substantial overdose, the possibility of inducing vomiting to eliminate the unabsorbed active ingredient may be considered.
05.0 PHARMACOLOGICAL PROPERTIES
05.1 Pharmacodynamic properties
Pharmacotherapeutic group: Drugs for the treatment of bone diseases - other drugs that affect bone structure and mineralization.
ATC code: M05BX03.
Mechanism of action
In vitro, strontium ranelate:
- increases bone formation in bone tissue cultures, as well as the replication of osteoblast precursors and collagen synthesis in bone cell cultures.
- decreases bone resorption by reducing osteoclast differentiation and their resorption activity.
This determines a rebalancing of bone turnover in favor of its formation.
The activity of strontium ranelate has been demonstrated in several experimental studies. In particular, in intact rats, strontium ranelate increases trabecular bone mass, number and thickness of trabeculae; this leads to an improvement in bone strength.
Strontium is mainly absorbed on the crystalline surface, and only to a limited extent does it replace calcium in the apatite crystal in newly formed bone in both animals and humans under treatment. Strontium ranelate does not change the characteristics of the bone crystal. In iliac crest bone biopsies taken after treatment with strontium ranelate 2 g / day for up to 60 months in phase III studies, no deleterious effects on bone quality or mineralization were observed.
The combined effects of the distribution of strontium in bone (see section 5.2) and the higher x-ray absorption of strontium compared to calcium, lead to an increase in the value of bone densitometry (BMD), measured by double-beam photon absorptiometry (DXA) . Available data indicate that these factors account for approximately 50% of the observed changes in BMD over 3 years of treatment with PROTELOS 2 g / day. This should be taken into consideration when evaluating changes in BMD during treatment with PROTELOS. In phase III studies, which demonstrated the efficacy of PROTELOS treatment in reducing fractures, PROTELOS increased mean BMD from inclusion by approximately 4% annually at lumbar vertebrae and 2% annually at lumbar vertebrae of the femoral neck, which reaches, depending on the study, respectively from 13 to 15% and from 5 to 6% after 3 years.
In phase III studies, compared to placebo, biochemical markers of bone formation (specific alkaline phosphatase and C-terminal propeptide of type I procollagen) were increased and those of bone resorption (serum C-telopeptide and urinary crosslinks) of N-telopeptide) decreased from the third month to the third year of treatment.
In addition to the primary pharmacological effects of strontium ranelate, slight decreases in serum calcium and parathyroid hormone (PTH) levels, increases in blood phosphorus concentration and total alkaline phosphatase activity have been observed, with no clinical consequences.
Clinical efficacy
Osteoporosis is defined as BMD of the spine or hip that is 2,5 or more standard deviations lower than the mean value in the normal young population. Some risk factors are associated with postmenopausal osteoporosis, including low bone mass, low bone mineral density, early menopause, smoking, and a family history of osteoporosis. The clinical consequence of osteoporosis is fractures. The risk of fractures increases as the number of risk factors increases.
Treatment of postmenopausal osteoporosis
The study program to evaluate fracture reduction with PROTELOS consisted of two phase III placebo-controlled studies: the SOTI study and the TROPOS study. The SOTI study involved 1.649 postmenopausal women with documented osteoporosis (low lumbar BMD and prevalent vertebral fractures) and an average age of 70 years. The TROPOS study involved 5.091 postmenopausal women with osteoporosis (low femoral neck BMD and at least one fracture in more than half of the patients) and a mean age of 77 years. Together, the SOTI and TROPOS studies enrolled 1.556 patients over the age of 80 at the time of inclusion (23,1% of the study population). In both studies, in addition to therapy (2 g / day of strontium ranelate or placebo), the patients took adequate calcium and vitamin D supplements.
PROTELOS reduced the relative risk of new vertebral fractures by 41% over 3 years of treatment in the SOTI study (table 1). The effect was significant from the first year. Similar benefits have been demonstrated in women with multiple fractures at enrollment. With respect to clinical vertebral fractures (defined as fractures associated with back pain and / or a decrease in body height of at least 1 cm), the relative risk was reduced by 38%. PROTELOS also reduced the number of patients with a reduction in body height by at least 1 cm compared to placebo. The quality of life assessment using the specific QUALIOST scale, as well as the general health perception scores of the general SF-36 scale, indicate the benefits of PROTELOS, compared to placebo.
The efficacy of PROTELOS in reducing the risk of new vertebral fractures was confirmed by the TROPOS study, even for osteoporotic patients without fragility fractures, at the time of inclusion.
| Table 1: Incidence of patients with vertebral fractures and relative risk reduction | |||
| About us | Placebo | PROTELOS | Relative risk reduction vs. placebo (95% CI), p-value |
| SOTI | N = 723 | N = 719 | |
| New vertebral fracture over 3 years | 32,8% | 20,9% | 41% (27-52), p |
| New vertebral fracture within 1 year | 11,8% | 6,1% | 49% (26-64), p |
| New clinical vertebral fracture over 3 years | 17,4% | 11,3% | 38% (17-53), p |
| TROPOS | N = 1.823 | N = 1.817 | |
| New vertebral fracture over 3 years | 20,0% | 12,5% | 39% (27-49), p |
A joint analysis of the SOTI and TROPOS studies showed that, in patients over 80 years of age at the time of inclusion, PROTELOS reduced the relative risk of new vertebral fractures by 32% over 3 years of treatment (incidence of 19,1 , 26,5% with strontium ranelate vs XNUMX% with placebo).
In an a-posterior analysis of patients from SOTI and TROPOS studies with lumbar vertebrae and / or femoral neck BMD in the range of osteopenia at the time of inclusion and with no prevalent fractures, but with at least one additional fracture risk factor (N = 176), PROTELOS reduced the risk of a first vertebral fracture by 72% over 3 years (incidence of vertebral fracture 3,6% with strontium ranelate vs 12,0% with placebo).
An a-posterior analysis was performed in a subgroup of TROPOS patients of particular medical interest and at high risk of fractures [defined as patients with a femoral neck BMD T-score ≤-3 SD (corresponding manufacturer range at -2,4 SD according to NHANES III) and an age ≥ 74 years (n = 1.977, ie 40% of the TROPOS study population)]. In this group, over 3 years of treatment, PROTELOS reduced the risk of hip fractures by 36% compared to placebo (table 2).
| Table 2: Incidence of patients with hip fractures and relative risk reduction in patients with BMD ≤-2,4 SD (NHANES III) and age ≥74 years | |||
| About us | Placebo | PROTELOS | Relative risk reduction vs. placebo (95% CI), p-value |
| TROPOS | N = 995 | N = 982 | |
| Hip fracture over 3 years | 6,4% | 4,3% | 36% (0-59), p=0,046 |
Treatment of osteoporosis in humans
The efficacy of PROTELOS was demonstrated in men with osteoporosis in a double-blind, placebo-controlled, 2-year study with a main analysis conducted after one year in 243 patients (Intention to treat population, 161 patients on treatment). with strontium ranelate) at high risk of fractures (mean age 72,7 years; mean lumbar BMD with a T-score of -2,6; 28% prevalent vertebral fracture).
All patients received daily calcium (1000 mg) and vitamin D (800 IU) supplements.
Statistically significant increases in BMD values were observed as early as 6 months after starting treatment with PROTELOS compared to placebo.
A statistically significant increase in mean lumbar spine BMD values was observed over 12 months, main efficacy criterion (E (SE) = 5,32% (0,75); 95% CI = [3,86 ; 6,79]: p menopause.
Statistically significant increases in femoral neck BMD and total femoral BMD values were observed (p
Pediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with PROTELOS in all subsets of the pediatric population in osteoporosis (see section 4.2 for information on pediatric use).
05.2 Pharmacokinetic properties
Strontium ranelate consists of 2 stable strontium atoms and a molecule of ranelic acid, an organic component that represents the best compromise, in terms of molecular weight, pharmacokinetics and acceptability of the medicinal product. The pharmacokinetics of strontium and ranelic acid were evaluated in healthy young male volunteers, in healthy postmenopausal women and, during long-term treatment, in men with osteoporosis and in women with postmenopausal osteoporosis, including elderly.
Absorption, distribution, plasma protein binding of ranelic acid is low due to its high polarity. There is no accumulation of ranelic acid and no evidence of metabolism in animals and humans. Absorbed ranelic acid is rapidly eliminated unchanged via the urine.
Absorption
The absolute bioavailability of strontium is 25% (range 19-27%) after an oral dose of 2 g of strontium ranelate. Maximum plasma concentrations are reached 3-5 hours after a single dose of 2 g.
Steady state is reached after 2 weeks of treatment. Intake of strontium ranelate with calcium or food reduces the bioavailability of strontium by approximately 60 - 70%, compared with administration 3 hours after a meal. Due to the relatively slow absorption of strontium, food and calcium intake before and after PROTELOS should be avoided. Oral vitamin D supplementation does not interfere with strontium exposure.
Distribution
Strontium has a volume of distribution of approximately 1 l / kg. The binding of strontium to human plasma proteins is low (25%) and strontium has a high affinity for bone tissue. Measurement of strontium concentrations in iliac crest bone biopsies from patients treated for up to 60 months with strontium ranelate 2 g / day shows that bone strontium concentrations can reach a plateau after approximately 3 years of treatment. There are no patient data demonstrating the kinetics of elimination of strontium from bone after discontinuation.
Biotrasformazione
As a divalent cation, strontium is not metabolized. Strontium ranelate does not inhibit the cytochrome P450 enzyme complex.
elimination
The elimination of strontium is independent of time and dose. The effective half-life of strontium is approximately 60 hours. Strontium excretion occurs through the kidneys and the gastrointestinal tract. Its plasma clearance is approximately 12 mL / min (CV 22%) and its renal clearance approximately 7 mL / min (CV 28%).
Pharmacokinetics in particular populations
Elderly patients
Population pharmacokinetic data showed no correlation between age and apparent clearance of strontium in the affected population.
Kidney failure
In patients with moderate to moderate renal impairment (creatinine clearance 30-70ml / min), strontium clearance decreases as creatinine clearance decreases (approximately 30% decrease in creatinine clearance range 30 to 70ml / min); this leads to an increase in plasma strontium levels. In phase III studies, 85% of patients had creatinine clearance between 30 and 70 mL / min, 6% less than 30 mL / min at inclusion, and mean creatinine clearance was 50 mL / min . Therefore, no dosage adjustment is required in patients with moderate to moderate renal impairment. There are no pharmacokinetic data in patients with severe renal impairment (creatinine clearance
Hepatic failure
There are no pharmacokinetic data in patients with hepatic insufficiency. Due to the pharmacokinetic properties of strontium, no effect is expected.
05.3 Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, genotoxicity, carcinogenic potential.
In rodents, chronic oral administration of high doses of strontium ranelate resulted in bone and tooth abnormalities, consisting mainly of spontaneous fractures and delayed mineralization, which were reversible after discontinuation of treatment. These effects were seen with a level of strontium in the bones 2 to 3 times higher than the levels found in humans after treatment lasting up to 3 years. Data on skeletal accumulation of strontium ranelate in long-term exposure are limited.
Developmental toxicity studies have resulted in bone and tooth abnormalities in the offspring of rats and rabbits (eg, bowing of long bones and wavy ribs). These effects are reversible 8 weeks after stopping treatment.
Environmental Risk Assessment (ERA)
The environmental risk assessment of strontium ranelate was conducted in accordance with the European guidelines relating to the ERA.
Strontium ranelate does not present a risk to the environment.
06.0 PHARMACEUTICAL INFORMATION
06.1 Excipients
Aspartame (E 951)
Maltodextrin
Mannitol (E 421)
06.2 Incompatibility
Not relevant.
06.3 Period of validity
- 3 years.
- Once reconstituted in water, the suspension is stable for 24 hours. However, it is recommended to drink the suspension immediately after preparation (see section 4.2).
06.4 Special precautions for storage
This medicine does not require any special storage conditions.
For storage conditions after reconstitution of the medicinal product, see section 6.3.
06.5 Nature of the immediate packaging and contents of the package
Paper / polyethylene / aluminum / polyethylene bags.
packs
Packs of 7, 14, 28, 56, 84 or 100 sachets.
Not all pack sizes may be marketed.
06.6 Instructions for use and handling
No special instructions.
07.0 MARKETING AUTHORIZATION HOLDER
SERVIER LABORATORIES
50, rue Carnot
92284 Suresnes cedex
France
08.0 MARKETING AUTHORIZATION NUMBER
EU/1/04/288/003
AIC n ° 036558031 / E - pack of 28 sachets
09.0 DATE OF FIRST AUTHORIZATION OR RENEWAL OF THE AUTHORIZATION
Date of first authorization: 21 September 2004
Date of most recent renewal: May 22, 2014
10.0 DATE OF REVISION OF THE TEXT
06/2014